Risikofaktor Altwerden |
||
Statistisch gesehen erkrankt jeder vierzehnte Mensch über 65 Jahre an Alzheimer. Und weil wir immer älter werden, wird es auch immer mehr Alzheimerpatienten geben. Die Grundlagenforschung versucht, dieser äusserst komplexen Krankheit mit Tiermodellen auf die Spur zu kommen. |
||
| Der Terminus «Alzheimer’sche Erkrankung» (AD) wurde vor rund neunzig Jahren von Emil Kraepelin eingeführt. Es handelt sich hierbei um eine progressiv verlaufende demenzielle Erkrankung, die derzeit weder signifikant verlangsamt noch gar gänzlich gestoppt werden kann. Den grössten Risikofaktor dieser verbreitetsten Form der Demenz stellt das Altwerden dar: Etwa 7% der Bevölkerung über 65 Jahren ist von Alzheimer betroffen und der Anteil an der Gesamtbevölkerung und die damit verbundenen immensen Kosten werden wachsen, da sich das Altersprofil immer mehr verschiebt. Während um 1900 nur 1% der Weltbevölkerung über 65 Jahre alt war, waren es 1992 schon über 6% und für das Jahr 2050 prognostiziert man bereits einen Anteil von 25%! Jedermann weiss, dass es bei Alzheimerpatienten zu immer offensichtlicheren Persönlichkeitsveränderungen kommt, die einhergehen mit einem fortschreitenden Verlust des Gedächtnisses. Fähigkeiten, die als Kleinkind erworben wurden, gehen in umgekehrter Reihenfolge unwiederbringlich verloren – für die Patienten selbst, aber auch für die Angehörigen eine äusserst schwie-rige Situation. Was aber geht in den Gehirnen der Alzheimerpatienten vor sich? Merkmale Die Alzheimerdemenz ist die Folge pathologischer Gewebeveränderungen im Gehirn, die zunächst in ganz bestimmten Hirnregionen auftreten und im Endstadium fast alle Hirnareale betreffen. In den degenerierenden Gehirnen finden sich zwei Kategorien dieser pathologischen Läsionen: stärkeförmige (amyloide Plaques) und neurofibrilläre Bündel. Amyloide Plaques sind im wesentlichen eine über Jahre und Jahrzehnte erfolgte Ablagerung des kurzen Ab-Peptides (ein Spaltprodukt des Amyloid Precursor Protein APP). Neurofibrillenbündel bestehen aus über- und abnormal phosphoryliertem Tau-Protein, das in Form von Paired Helical Filaments (PHFs) aggregiert. Die Phosphorylierung ist ei-ner der wichtigsten Regulationsmechanismen unseres Körpers, da über die Anhängung von Phosphatgruppen an bestimmte Aminosäurereste unter anderem Enzymaktivitäten oder die Bindung zwischen zwei Proteinen reguliert werden. Amyloide Plaques findet man extrazellulär, neurofibrilläre Bündel in ihrer markantesten Ausprägung intrazellulär, in Form von «Knäueln», sogenannten «Tangles». Beide Läsionen sind anatomisch separiert. Bestimmte neuronale Populationen sind betroffen, andere nicht. Ein weiteres Merkmal der Alzheimerschen Erkrankung ist der massive neuronale Zelltod, der in bestimmten Hirnregionen bis zu 90% aller Nervenzellen betreffen kann. Warum gerade bestimmte Neuronen Ablagerungen aufweisen und/oder absterben, weiss man nicht. Ein gewaltiger Fortschritt in der Alzheimerforschung wurde erzielt, als erste Gene identifiziert und kloniert werden konnten, die mit der Erkrankung in familiär auftretenden Formen segregieren. Familiäre Formen machen etwa 10% aller Fälle aus, sporadische 90%. Mutationen wurden auf den Chromosomen 1 und 14 gefunden sowie auf Chromosom 21, auf dem sich auch das Gen für das APP-Protein befindet. Diese Mutationen sind dominant und führen bei den betroffenen Patienten zu einem frühen Einsetzen der Krankheit (Early Onset AD). Ein viertes Gen, ApoE, kommt in der Bevölkerung in drei Varianten vor und erhöht bei Vorliegen einer bestimmten allelen Form die Wahrscheinlichkeit, an AD zu erkranken (Late Onset AD). Es kann mit Sicherheit davon ausgegangen werden, dass noch weitere Alzheimergene gefunden werden. Kopplungsanalysen weisen beispielsweise auf Gene auf den Chromosomen 3 und 12 hin. Umfassendes Tiermodell gesucht Es ist offensichtlich, dass man gerade bei Krankheiten wie AD, die komplexe Systeme wie das Gehirn betreffen, auf Tiermodelle angewiesen ist und sich nicht auf Zellkulturexperimente beschränken kann. Was steht an Tiermodellen zur Verfügung? Bei alten Affen findet man amyloide Plaques, aber keine neurofibrillären Läsionen. Bei Wiederkäuern finden sich Strukturen, die Tangles ähneln, aber keine Plaques. Alte Polarbären wiederum weisen Plaques auf, aber keine Tangles, wenn auch Zytoskelettveränderungen, die ähnlich auch bei AD zu beobachten sind. Abgesehen davon, dass alle diese Modelle nicht «vollständig» sind, ist ein Polarbär auch nicht gerade ein praktisches Versuchstier. Das Tiermodell der Wahl ist die Maus, die auch wir als Versuchstier verwenden. Mäuse haben eine kurze Generationszeit, lassen sich leicht züchten, sind genetisch gut charakterisiert, und ihr Erbgut lässt sich manipulieren, indem man fremde Gene einschleust (das heisst: klassische transgene Mäuse generiert), Gene ausschaltet (sogenannte Knockouts) oder Gene in situ mutiert (sogenannte Knockins). Unser Fernziel ist, ein umfassendes transgenes Tiermodell für die Alzheimersche Erkrankung zu erhalten, welches Amyloidablagerung, neurofibrilläre Läsionen und neuronalen Zelltod einschliesst, um so einen Einblick in den Verlauf der pathogenetischen Kaskade und die räumliche und zeitliche Korrelation der einzelnen Läsionen zueinander zu erhalten. Andererseits erhoffen wir uns von einem umfassenden Tiermodell entscheidende Hinweise auf mögliche neue Drug targets. Finden wir beispielsweise heraus, dass die erhöhte Aktivität eines Proteins im Tiermodell zu einer Alzheimerpathologie führt, dann werden wir in einem einfachen Testverfahren versuchen, die Aktivität dieses Proteins zu messen. In einem Massenscreening wird man Substanzen finden, welche spezifisch die Aktivität des betreffenden Proteins reduzieren. Diese Substanzen kann man dann direkt im Tiermodell auf ihre Wirksamkeit in bezug auf die pathologischen Läsionen testen, bevor sie an Alzheimerpatienten eingesetzt werden. In den letzten Jahren wurden in verschiedenen Laboratorien einige Versuche unternommen, humanes APP (mit krankheitsverursachenden Mutationen) in transgenen Tieren zu exprimieren, um so ein Tiermodell für die Alzheimersche Erkrankung zu erhalten. Die besten Modelle weisen für Alzheimer charakteristische Aß-enthaltende Plaques auf und zeigen auch, dass offensichtlich ein bestimmter Aß-Schwellenwert überschritten werden muss, um Plaques zu erhalten. Während die vorgestellten Modelle ein Charakteristikum der AD-Pathologie, nämlich die Bildung amyloider Plaques, experimentell nachbildeten, erzeugten diese Modelle weder neurofibrilläre Läsionen (mit Paired Helical Filaments) noch massiven neuronalen Zellverlust. Es handelt sich hier deshalb eigentlich um Hirnamyloidosenmodelle. Es ist anzumerken, dass der Schweregrad der Demenz in AD-Patienten am besten mit dem Verlust an Synapsen korreliert (Synapsen sind Fortsätze, über die eine Nervenzelle den Reiz weiterleitet), und nicht mit der Anzahl amyloider Plaques. Keines der bislang publizierten Modelle weist jedoch Synapsenverlust auf.
Das Tau-Modell Wir haben uns vor einigen Jahren zunächst der Tau-Seite zugewandt und Mäuse hergestellt, die verschiedene potentiell interessante Kinasen (das sind Enzyme, die Proteine wie das Tau phosphorylieren können) im Gehirn überexprimieren, haben jedoch keine Veränderung in der Tau-Phosphorylierung beobachten können. Man weiss, dass Down-Syndrom-Patienten (die drei Kopien des APP kodierenden Chromosoms 21 besitzen) recht früh (im Alter von 35 Jahren) eine Alzheimer-Neuropathologie entwickeln. Interessanterweise ist die Expression der Tau-RNA beim Down-Syndrom erhöht. Wir haben deshalb transgene Mäuse hergestellt, die humanes Tau im Gehirn überexprimieren. Zu unserer Freude fanden wir, wie bei AD, neben einer Tau-Expression im Axon, wo das Tau-Protein normalerweise lokalisiert ist, eine starke Lokalisation im Zellkörper und in den Dendriten. Ausserdem ist Tau an mehreren diagnostischen Aminosäurere-sten phosphoryliert, die auch bei AD phosphoryliert sind. Eine zweite Generation tau-transgener Mäuse mit fünffach stärkerer transgener Tau-Expression zeigt neben der Tau-Pathologie auch noch eine kugelige Ablagerung der Neurofilament-Proteine, ähnlich, wie man sie bei Lewykörper-Erkrankungen findet. (Lewykörper findet man beispielsweise bei der Parkinsonschen Erkrankung.) Bei keinem der Mausmodelle findet man jedoch Plaques oder neurofibrilläre Läsionen, es handelt sich beim Tau-Modell deshalb um ein Modell für ein Frühstadium der Alzheimererkrankung. Unlängst sind auch Mutationen im Tau-Gen beschrieben worden, die bei Menschen zu einer alzheimerähnlichen Demenz führen. Wir versuchen, diese Mutationen in transgenen Mäusen zu exprimieren.
Weiterführende Ansätze In einem nächsten Schritt haben wir uns einer Phosphatase zugewandt, die möglicherweise reduzierte Aktivität bei Alzheimer zeigt, der Phosphatase 2A. Ein kompletter Knockout der katalytischen Untereinheit dieser Phosphatase führt zu embryonaler Letalität, schon wenn sich die dritte Keimschicht des Embryos, das Mesoderm, ausbildet. Um embryonale Letalität zu umgehen, arbeiten wir an einem gewebespezifischen Knockout, bei dem die Phosphatase 2A nur in Neuronen ausgeschaltet wird, und zwar erst dann, wenn die Neuronen postmitotisch werden, das heisst, sich nicht mehr weiterteilen. In einem parallelen Ansatz versuchen wir, eine dominant negativ wirkende Form der katalytischen Untereinheit in Neuronen in hohen Mengen zu exprimieren. Die Idee dahinter ist, dass die mutante Form als überwiegende die funktionale, endogen vorkommende von Bindungsstellen verdrängt und somit die Enzymaktivität unterbindet. In Zusammenarbeit mit der pharmazeutischen Industrie sind wir dabei, ein modales transgenes System zu entwickeln, das durch entsprechende Kombination pathologischer Läsionen via Kreuzung mehrerer Tiermodelle zu einem umfassenden Tiermodell für die AD führt. Unsere Grundlagenforschung – angesiedelt an der Abteilung für Psychiatrische Forschung und assoziiert mit der Psychiatrischen Universitätsklinik – bildet eine Schnittstelle zwischen klinischer und Grundlagenforschung. Wir hoffen so, einen Beitrag zur Bekämpfung der Alzheimerschen Erkrankung leisten zu können. |
Dr. Jürgen Götz ist Oberassistent am Institut für Molekulare Psychiatrie der Universität Zürich.
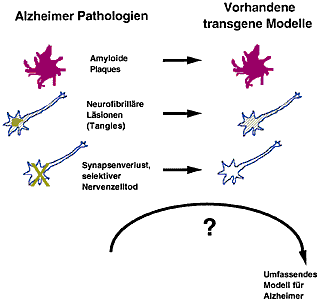 Abbildung 1: Drei histopathologische Veränderungen bei Alzheimer (linke Spalte) und inwieweit diese Veränderungen in publizierten transgenen Modellen reproduziert werden konnten (rechte Spalte).
Abbildung 1: Drei histopathologische Veränderungen bei Alzheimer (linke Spalte) und inwieweit diese Veränderungen in publizierten transgenen Modellen reproduziert werden konnten (rechte Spalte).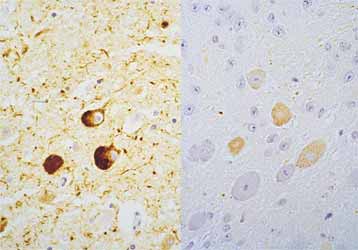 Abbildung 2: Anfärbung eines Schnittes durch ein menschliches Alzheimergehirn (links) und ein transgenes Maushirn (rechts) mit einem Antikörper, der ein Tau-Protein erkennt, das an einer ganz bestimmten Stelle eine Phosphatgruppe trägt.
Abbildung 2: Anfärbung eines Schnittes durch ein menschliches Alzheimergehirn (links) und ein transgenes Maushirn (rechts) mit einem Antikörper, der ein Tau-Protein erkennt, das an einer ganz bestimmten Stelle eine Phosphatgruppe trägt.