VON CHARLES WEISSMANN
Prionen sind Erreger, die bei Rindern Rinderwahnsinn, bei Schafen Scrapie und bei Menschen die Creutzfeldt-Jakob-Erkrankung sowie Kuru verursachen. Das Prion entsteht durch Verwandlung eines bestimmten Proteins.
[ Diagramm ]
Bis vor wenigen Jahren bestand kein Zweifel, dass alleübertragbaren Krankheiten entweder auf Viren oder auf zelluläre Organismen wie Bakterien, Protozoen oder vielzellige Parasiten zurückzuführen seien. Allen diesen Erregern ist gemeinsam, dass sie eigenes Erbmaterial, DNA oder RNA, enthalten und ihre Wesenszüge weitgehend eigenständig bestimmen. In letzter Zeit setzt sich jedoch die Meinung durch, dass bestimmte Erkrankungen des Zentralnervensystems, die «übertragbaren schwammartigen Enzephalopathien», auf einen andersartigen Erreger, Prion genannt, zurückzuführen sind, der sich von einem integralen Bestandteil des befallenen Organismus selbst ableitet, nämlich von einem Eiweiss (Protein), das vor allem im Hirn vorkommt. Im Gegensatz zuüblichen Erregern ist er ungemein widerstandsfähig gegenübliche Sterilisationsverfahren, und die Inkubationszeit dauert Monate bis Jahre.
Eine als «Scrapie» oder «Traberkrankheit» genannte Krankheit des Schafes wurde in den letzten Jahrzehnten als Prototyp von Prionenkrankheiten des Zentralnervensystems von Mensch und Tier erforscht. In den letzten Jahren ist vor allem in England eine neue Prionenerkrankung der Tiere epidemieartig aufgetreten, der Rinderwahnsinn. Man führt ihre Verbreitung auf die Verfütterung von aufgearbeiteten Schlachtabfällen zurück, in denen sich Überreste von mit Scrapie infizierten Tieren befanden. Obwohl ausgedehnte Untersuchungen keine Anhaltspunkte für eine Übertragung von Scrapie vom Schaf auf den Menschen ergaben, ist die Situation im Falle des Rinderwahnsinns noch unklar. Prionenerkrankungen des Menschen sind u. a. die Creutzfeldt-Jakob-Erkrankung (CJD) und Kuru. CJD tritt jährlich nur bei einem von einer Million Menschen auf. Kuru hingegen nahm in den ersten Jahrzehnten dieses Jahrhunderts in Papua-Neuguinea epidemische Ausmasse an. Es scheint, dass Kuru durch rituellen Kannibalismusübertragen wurde und die Epidemie im Verzehren von Leichenteilen eines an CJD erkrankten Individuums ihren Ursprung gehabt haben könnte.
Prionenerkrankungen des Menschen sind insofern ungewöhnlich, als sie einerseits ohne erkennbare Ursache in der allgemeinen Bevölkerung, anderseits gehäuft in bestimmten Familien auftreten. Die Krankheit ist in beiden Fällen experimentell auf das Tier und (ungewollt) auf den Menschen übertragbar. Indirekte Übertragung von Mensch zu Mensch kann zum Beispiel bei Transplantation menschlicher Gewebe oder Verabreichung von aus menschlichen Drüsen gewonnenen Hormonen erfolgen, vermutlich ausgehend von Spendern, die noch nicht erkennbar krank waren, aber den Erreger schon enthielten. Obwohl die Inkubationszeit gewöhnlich Jahre bis Jahrzehnte beträgt, kann die Krankheit nach Auftreten der ersten Symptome innerhalb weniger Monate zum Tod führen. Erste Krankheitszeichen beim Menschen sind Gedächtnis-, Schlaf- oder Bewegungsstörungen; die Krankheit führt zum Erlöschen geistiger Präsenz und endet, meistens innert Monaten nach den ersten Symptomen, unweigerlich mit dem Tod. Im Hirn findet man eine löchrige Degeneration der Hirnsubstanz und Anreicherung eines Eiweisses, PrPSc genannt, zum Teil in Form unlöslicher Ablagerungen, dem Amyloid. Es finden sich keine Anzeichen einer Entzündung oder Immunreaktion, wahrscheinlich weil der Erreger eine körpereigene Substanz ist.
Die von J. S. Griffith 1967 umrissene, später von S. Prusiner in detaillierter Form gefasste, «nur-Eiweiss»-Hypothese geniesst heute weite Akzeptanz. Sie besagt, dass das Prion keine Nukleinsäure enthält und identisch ist mit PrPSc, einer modifizierten Form von PrPC. PrPC ist ein normales Protein, das im nicht infizierten Wirtsorganismus vorkommt, vor allem an der Oberfläche von Neuronen. Wie unsere Arbeitsgruppe zeigte, ist die für PrP-Protein kodierende Information in einem wirtsspezifischen Gen enthalten. Nach Infektion mit Prionen findet die Umwandlung von PrPC in PrPSc statt. Bisher wurden keine chemischen Unterschiede zwischen den beiden Formen von PrP gefunden, wohl aber Unterschiede in der Faltung, das heisst der räumlichen Anordnung der Eiweisskette. Demnach soll der Übergang von PrPC in PrPSc auf einer Veränderung der Faltung des Moleküls beruhen, die durch PrPSc gesteuert wird (siehe Abbildung) und kaskadenartig abläuft.
Obwohl menschliche Prionenerkrankungen selten sind und die meisten sporadischen Fälle ohne nachweisbaren Zusammenhang mit äussern Umständen auftreten, so gibt es Familien, in denen etwa die Hälfte der Mitglieder im fortschreitenden Alter an CJD erkranken. Das Erbmaterial dieser Patienten weist immer eine Mutation, das heisst eine Veränderung im PrP-Gen, auf, die zu einer Strukturänderung des PrP-Proteins führt. Im Rahmen der «nur-Eiweiss»-Hypothese könnte das bedeuten, dass solche Mutationen die spontane Umfaltung von PrPC zu PrPSc, die normalerweise enorm selten ist, erleichtern und damit die Wahrscheinlichkeit der Umwandlung so weit erhöhen, dass die Krankheit während der Lebensspanne des Betroffenenen auftritt.
Die «nur-Eiweiss»-Hypothese sagt voraus, dass Tiere ohne PrP resistent gegen Scrapie sein sollten. Ferner dürfte sich der Erreger in ihnen nicht mehr vermehren können. Um diese Voraussagen zuüberprüfen, hat unsere Arbeitsgruppe durch gentechnische Methoden eine Mauslinie erzeugt, in der das PrP-Gen zerstört worden war. In den Gehirnen solcher Tiere konnte kein PrP mehr nachgewiesen werden. Mit Prionen inokulierte, PrP-lose Mäuse waren vollständig resistent gegen Scrapie und blieben zeitlebens gesund, während normale Mäuse 180 Tage nach der Inokulation erkrankten und kurz danach eingingen. Um die Vermehrung von Prionen zu untersuchen, haben wir zu verschiedenen Zeiten nach der Inokulation mit Scrapie den Gehalt des Hirns an Prionen bestimmt. In normalen Mäusen konnte Vermehrung des Erregers im Hirn spätestens nach acht Wochen festgestellt werden, und nach zwanzig Wochen war der Titer auf fast eine Milliarde Prionen pro Gramm Hirn angestiegen. Eine Vermehrung von Prionen in PrP-losen Tieren fand hingegen nicht statt. Um uns zu vergewissern, dass die Resistenz PrP-loser Mäuse gegen Scrapie in der Tat auf der Abwesenheit dieses Eiweisses beruhte, haben wir in diese Mauslinie ein oder mehrere Gene für PrP durch gentechnische Methoden eingeführt. Wir fanden, dass solche transgene Mäuse nicht nur ihre Anfälligkeit für Scrapie zurückgewannen, sondern, wenn mehrere Genkopien eingeführt wurden und es zu einer Überproduktion von PrP kam, die Anfälligkeit stark gesteigert wurde, was sich in einer auf bis zu 60 Tagen verkürzten Inkubationszeitäusserte.
Die hier beschriebenen Befunde sprechen stark dafür, dass das Prion teilweise oder ganz aus PrPSc besteht und dass es keiner Nukleinsäure als essentiellem Bestandteil bedarf.
Weil Mäuse mit zerstörten PrP-Genen resistent sind gegen Scrapie, sich aber sonst nicht von normalen Tieren unterscheiden, sollte es möglich sein, mittels PrP-Gen-Inaktivierung Rinder und Schafe zu züchten, welche gegen BSE beziehungsweise Scrapie resistent sind; die technischen Voraussetzungen für das Verfahren bei diesen Tieren sind allerdings noch nicht erarbeitet. Eine Reduktion der PrPC-Synthese, welche durch Verabreichung eines noch zu entdeckenden, die Expression des PrP-Gens spezifisch hemmenden Wirkstoffes zu erzielen wäre, könnte das Fortschreiten früherfasster Prionenerkrankungen verzögern.
Schliesslich geht aus meinen Ausführungen hervor, welchüberragende Rolle der Erzeugung und Untersuchung genetisch veränderter Tiere zukommt. Ohne die Möglichkeit, genetisch veränderte Organismen herzustellen, würde die moderne medizinische und biologische Forschung weitgehend unterbunden und unser Land in die wissenschaftliche Drittwelt zurückgestossen.
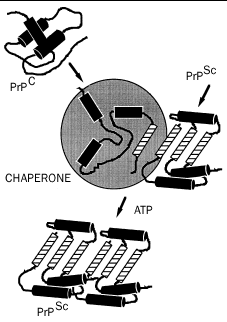 Ein Modell für die katalysierte konformationelle Umwandlung von PrPC in PrPSc.
Ein Modell für die katalysierte konformationelle Umwandlung von PrPC in PrPSc.
PrPC (a) wird partiell entfaltet (b), vielleicht mit Hilfe eines biochemischen Katalysators, Chaperon genannt, und unter dem Einfluss des als Matrize dienenden PrPSc zu einem diesem ähnelnden Molekül aufgefaltet (c).
Dr. Charles Weissmann
(weissma@molbio1.unizh.ch) ist ordentlicher Professor und Direktor des Instituts für Molekularbiologie der Universität Zürich.
![]()
unipressedienst Pressestelle der Universität Zürich
Felix Mäder (fmaeder@zuv.unizh.ch)
http://www.unizh.ch/upd/magazin/1-96/prionen.html
Last update: 1-APR-96