Leitungsstörungen |
||
Wie entwickelt sich das periphere Nervensystem, wie funktioniert es, und welche molekularen Grundlagen können Erkrankungen dieses Systems erklären? Eine Arbeitsgruppe am Institut für Zellbiologie der ETH Zürich sucht nach Antworten. |
||
Periphere Nerven bestehen hauptsächlich aus langen Fortsätzen von Neuronen, den Axonen, welche unsere Muskeln und die Haut mit der Steuerzentrale im Rückenmark und Gehirn verbinden. Eine reibungslose Funktion dieser Leitungen ist für unseren Körper lebensnotwendig, da auf diese Weise die korrekte Übertragung von Steuerimpulsen zwischen der Peripherie und dem Zentralnervensystem gewährleistet wird. Eine der wichtigsten Herausforderungen für dieses System besteht darin, dass elektrische Signale oft eine sehr lange Strecke entlang eines Nervs mit grosser Geschwindigkeit zurücklegen müssen. Damit ein solcher elektrischer Impuls auf seinem Weg entlang des Nervs nicht zu stark abgeschwächt wird (oder sogar verlorengeht), sind die einzelnen Nervenfasern ähnlich wie ein elektrisches Kabel gebaut. Die Axone werden dabei von Glialzellen unterstützt. Diese sogenannten Schwannschen Zellen lagern sich entlang der Axone ab und winden ihre Zellmembran mehrfach um die neuronalen Fortsätze. Da die Zellmembran mehrheitlich aus Lipiden und einigen wenigen, funktionell äusserst wichtigen Proteinen aufgebaut ist, entsteht so um das Axon eine kompakte und isolierende Fettschichthülle, die Myelin genannt wird. Molekularer Dialog Der Prozess der Myelinisierung während der Entwicklung oder Regeneration eines peripheren Nervs ist von einem intensiven Zusammenspiel zwischen Neuronen und Schwannschen Zellen geprägt. Dabei spielen direkte Kontakte über Membranproteine und der Austausch von löslichen Wachstumsfaktoren eine entscheidende Rolle. Unter anderem steuern diese Interaktionen die korrekte Positionierung von Axonen und Schwannschen Zellen innerhalb des Nervs, die zeitlich und örtlich korrekte Initiation der Myelinisierung, deren Fortführung sowie das Ende des Myelinisierungsprozesses. Die kritischen Wechselwirkungen zwischen Neuronen und Schwannschen Zellen sind jedoch nicht auf die Regulation der Myelinisierung beschränkt. Neuere Studien, unter anderem auch aus unserer Arbeitsgruppe, zeigen, dass schon während der frühen Entwicklung ein kritischer molekularer Dialog zwischen den beiden Zelltypen stattfindet. Am eindrücklichsten wird dies durch spezielle Mäuse belegt, welche auf Grund einer gezielten genetischen Veränderung keine Schwannschen Zellen mehr ausbilden. In diesen Tieren werden die normalerweise mit den Schwannschen Zellen assoziierten Neuronen stark geschädigt und sterben. Auf der anderen Seite können auch Schwannsche Zellen ohne Axonen nicht korrekt ausreifen und sind nicht überlebensfähig. Die «Trembler»-Maus Auf diesem Hintergrund erstaunt es nicht, dass ein gestörtes Verhältnis zwischen Schwannschen Zellen und Neuronen auch Krankheiten verursachen kann. Anfang der neunziger Jahre, während eines durch den Schweizerischen Nationalfonds unterstützten Forschungsaufenthaltes im Labor von Prof. Eric Shooter an der Stanford University in Kalifornien, studierten wir mit Hilfe eines molekularbiologischen Screenings, welche Moleküle bei der Regeneration von peripheren Nerven eine wichtige Rolle spielen könnten. Eines der so gefundenen Proteine stellte sich als ein Bestandteil von Myelin heraus und wurde von uns auf Grund seines Molekulargewichtes von 22 Kilodalton «Peripheres Myelin-Protein 22» (kurz PMP22) genannt. Angeregt durch diese Daten erinnerten wir uns an ältere Studien, welche eine spontane Mausmutante beschreiben, die ein aufmerksamer Tierwärter Anfang der fünfziger Jahre in England zufällig in seiner Mauszucht entdeckt hatte. Ihm war eine Maus aufgefallen, die sich langsamer als ihre Geschwister bewegte, leichte Lähmungen der Hinterbeine zeigte und auch leicht zitterte. Diese spezifischen Eigenschaften wurden dominant vererbt, das heisst, bei der Kreuzung einer affektierten Maus mit einer normalen Maus ist die Hälfte der Nachkommen wiederum von der Krankheit betroffen. Interessanterweise zeigten die peripheren Nerven von «Trembler»-Mäusen (wie dieser neue Stamm auf Grund des auffälligen Zitterns der Tiere genannt wurde) markante Myelinstörungen. Wir stellten nun die Hypothese auf, dass möglicherweise eine Mutation in dem von uns gefundenen PMP22-Gen die Eigenschaften der «Trembler»-Maus erklären könnte. Tatsächlich zeigten unsere Experimente, dass die Veränderung einer einzigen Aminosäure im PMP22-Protein (ausgelöst durch eine sogenannte Punktmutation im entsprechenden PMP22-Gen) mit dem «Trembler»-Phänotyp vererbt wird. Diese Entdeckung weckte unser Interesse endgültig. Mehrere Neurologen hatten nämlich darauf hingewiesen, dass die Pathologie in der «Trembler»-Maus eine verdächtige Ähnlichkeit zu den häufigsten Formen von erblichen peripheren Neuropathien im Menschen aufweist. Charcot-Marie-Toothsche Krankheit Diese Gruppe von Krankheiten, die weltweit bei einer von viertausend Personen vorkommt wurde Ende des letzten Jahrhunderts durch die französischen Ärzte Jean-Marie Charcot und Pierre Marie sowie durch ihren englischen Kollegen Howard Henry Tooth entdeckt. Sie beschrieben Patienten mit schweren Muskelschwächen und sensorischen Störungen in den Beinen und Füssen und zum Teil auch in den Händen. Man spricht deshalb von der Charcot-Marie-Toothschen Erkrankung (kurz CMT). Es gelang uns in der Folge in Zusammenarbeit mit den Genetikprofessoren Jim Lupski und Pragna Patel am Baylor College in Houston zu zeigen, dass die weitaus häufigste Form von CMT (CMT Typ 1A; CMT1A) tatsächlich zusammen mit einer Mutation vererbt wird, die das PMP22-Gen betrifft. Bei diesen Patienten ist die Leitfähigkeit peripherer Nerven durch eine chronische Schädigung des Myelins stark verringert, ein Umstand, der zu den beschriebenen klinischen Symptomen führt. Merkwürdigerweise findet man jedoch bei fast allen CMT1A-Patienten das PMP22-Gen selbst unverändert. Vielmehr tragen diese Personen in ihrem Erbgut statt der üblichen zwei Kopien des PMP22-Gens (das heisst je ein Gen auf dem mütterlich und väterlich vererbten Chromosom) drei Kopien des PMP22-Gens (sowie einiger benachbarter Gene auf Chromosom 17; in der Fachsprache die Folge einer ursprünglichen intrachromosomalen Duplikation während der frühen Meiose). Daraus ergab sich die Hypothese, dass ein Gendosis-Effekt, verursacht durch die zusätzliche Kopie des PMP22-Gens, für die Krankheit verantwortlich sein müsse. Tiermodelle Um diese Vermutung zu beweisen, führten wir in unserer Arbeitsgruppe an der ETH Zürich zusätzliche Kopien des PMP22-Gens in das Erbgut von Mäusen und in Zusammenarbeit mit Prof. Klaus-Armin Nave am Zentrum für Molekularbiologie in Heidelberg auch von Ratten ein. Wie erwartet, zeigten diese transgenen Tiere ein ähnliches Verhalten wie «Trembler»-Mäuse und pathologische und elektrophysiologische Veränderungen, wie sie bei CMT1A-Patienten gefunden werden. Eine Analyse der Krankheitsmechanismen in diesen Tiermodellen zeigte auf, dass bei CMT1A dem gestörten Dialog zwischen Neuronen und Schwannschen Zellen eine Schlüsselrolle zukommt. So sind nicht nur die myelinisierenden Zellen bei CMT1A betroffen, sondern auch die assoziierten Axone zeigen signifikante Schädigungen. Unsere bisherigen Daten lassen darauf schliessen, dass wir durch die Aufklärung der molekularen Mechanismen dieses Prozesses weitere grundlegende Erkenntnisse über das Zusammenwirken von Neuronen und Schwannschen Zellen gewinnen und damit auch CMT1A besser verstehen lernen können. Fernziel Therapie Die in diesem Projekt bisher gewonnenen Erkenntnisse haben zur Entdeckung der grundlegenden molekulargenetischen Defekte in verschiedenen Formen von CMT und der Bereitstellung von entsprechenden Tiermodellen geführt. Dadurch wurde auch ein direkter Fortschritt in der Klinik erreicht, indem CMT heute durch molekulargenetische Tests eindeutig pränatal und postnatal diagnostiziert werden kann. Somit konnte zum Beispiel die Frequenz der Entnahme von Nervenbiopsien drastisch verringert werden. Leider kennen wir heute jedoch noch keine Therapien oder Heilungsmöglichkeiten für CMT. Es ist deshalb eine wichtige Aufgabe unserer Forschung, die nun vorhandenen Tiermodelle im Bezug auf die involvierten Krankheitsmechanismen detailliert zu untersuchen mit dem Fernziel der Entwicklung einer rationalen Therapie für schwer betroffene Patienten. |
Dr. Ulrich Suter ist ausserordentlicher Professor für Zellbiologie am Institut für Zellbiologie der ETH Zürich.
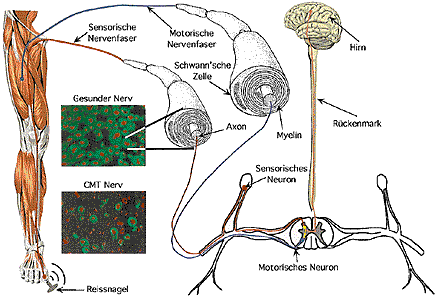 CMT affektierte motorische und sensorische Nerven: Die Einschübe zeigen Querschnitte durch den Nervenstrang eines gesunden und eines CMT-affektierten Nervs. Die Axone sind rot eingefärbt, und das Myelin erscheint grün. Starke Abnahme von myelinisierten Fasern in CMT-Nerven.
CMT affektierte motorische und sensorische Nerven: Die Einschübe zeigen Querschnitte durch den Nervenstrang eines gesunden und eines CMT-affektierten Nervs. Die Axone sind rot eingefärbt, und das Myelin erscheint grün. Starke Abnahme von myelinisierten Fasern in CMT-Nerven.